Non-coding region variants have an important role in developmental disorders
Today sees the publication of our latest paper in the American Journal of Human Genetics (AJHG). Read it here. The aim of this blog post is to share why we are so excited about this work and to outline a few of the key take home messages.
Some background - developmental disorders and the DDD project
Developmental disorders are severe conditions affecting normal childhood development. The majority of these conditions are caused by genetic variants (often altering only a single DNA base out of over 3 billion in each of our genomes!) that lead to changes in levels and/or function of important proteins. Whilst in some cases a child’s condition can be diagnosed using clinical measures alone, for many, a ‘genetic diagnosis’ is the first clue into what is happening, and how it can be most effectively treated. Knowing the genetic cause of the disorder also allows us to predict whether additional siblings will be at risk of the same condition, with important implications for counselling.
Given the huge advantages to knowing the genetic cause, the majority of patients with developmental disorders will undergo genetic testing as part of their clinical care. However, a genetic diagnosis is unfortunately uncovered in fewer than half of cases.
The Deciphering Developmental Disorders (DDD) project was first launched in 2011 with the aim of recruiting patients that do not have a genetic diagnosis. The study successfully collected data (both genetic sequencing data and detailed clinical phenotype data) for over 13,500 such individuals, and their parents. The project has two key goals: (1) to uncover the genetic variants causing disease in these patients, and (2) to learn more about the genetic basis of developmental disorders more broadly.
Current clinical genetic testing is mostly limited to the protein-coding genome
As you can imagine, ten years into the project, the DDD cohort has been pretty extensively studied. The vast majority of these analyses, however, have focussed on only a small proportion of the human genome; the ~1.5% that directly encodes proteins. These protein-coding regions are also the only regions that are usually assessed during clinical genetic testing. Limiting to these regions is a strategy that makes a lot of sense, given that variants that directly change the protein sequence are likely to have the largest impact. Only assessing a small portion of the genome also dramatically decreases both genetic sequencing costs and analysis time, making these tests more feasible in a healthcare setting.
But we are interested in looking for variants outside of these protein-coding regions (in the so-called ‘non-coding’ genome) that cause disease. We hope that by doing so, we can increase the number of patients getting a crucial genetic diagnosis.
In this work, we searched for variants in the DDD cohort in regions of the genome that are immediately adjacent to protein-coding regions and that are known to have very important roles in regulating the amount of protein that is produced (termed untranslated regions, or UTRs). We focussed on de novo variants, which are those that are detected in an affected child, but not in their healthy parents. These variants are infrequent, but have a higher likelihood of causing disease. We also only assessed variants in genes with already known links to developmental disorders; where protein-coding variants that lead to a reduction in levels or function of the protein are known to cause disease.
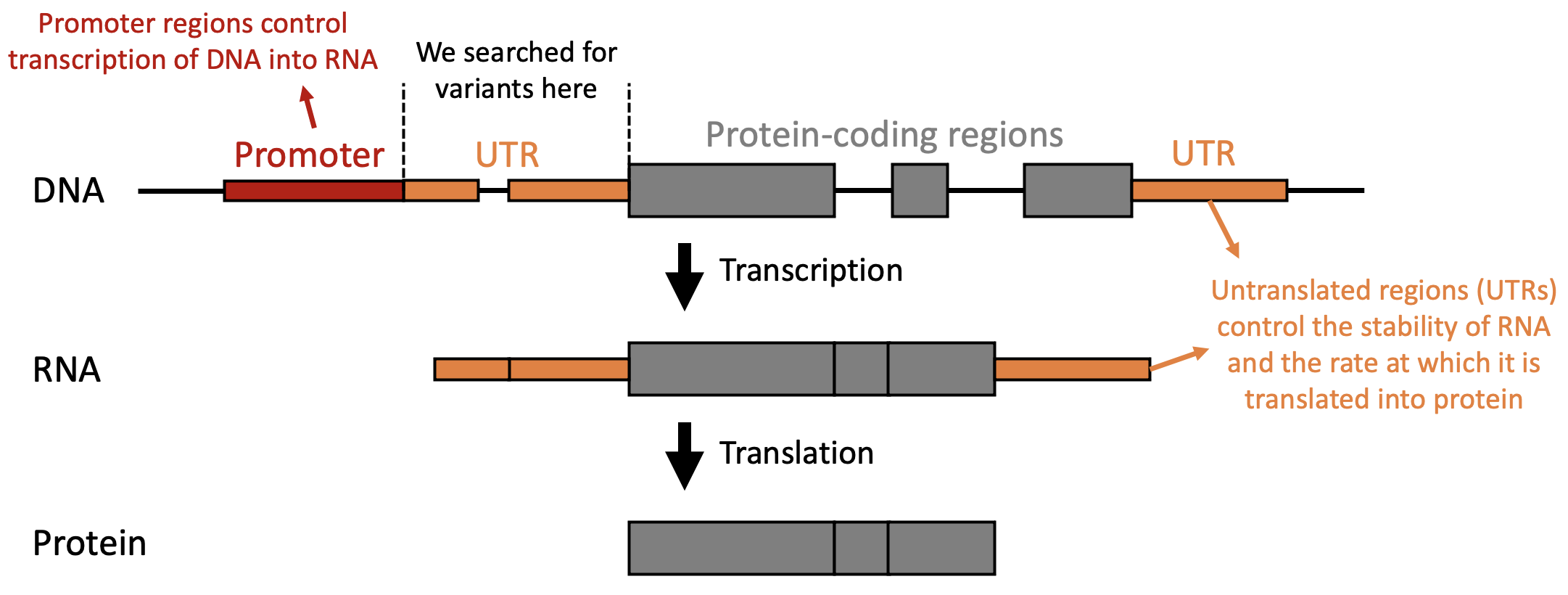
We identified variants in UTRs, which are regulatory elements that influence the amount of protein that is produced.
So what were our key findings?
New diagnoses for rare disease patients
We uncovered variants in seven individuals from the DDD cohort; two of these variants we then also found in four additional patients from other patient cohorts. We were able to return these diagnoses to the patients, finally giving them and their families answers and ending their long ‘diagnostic odyssey’.
New mechanisms underlying disease
Six of the variants we found impacted only one gene - MEF2C. We used both computational and laboratory based techniques to look into how these variants cause disease and found three different mechanisms:
Deleting promoter regions which are required to drive gene expression, resulting in lower levels of RNA transcript.
Reducing translation of RNA into protein, resulting in lower levels of MEF2C protein.
Disrupting MEF2C protein function, by preventing MEF2C binding to DNA.
All of these mechanisms ultimately result in lower levels of functional MEF2C protein.
Non-coding region variants are an important cause of rare disease
A quite surprising finding was that together, these non-coding region variants comprised almost ¼ of all genetic diagnoses identified in MEF2C in DDD. All of these would be missed in standard clinical genetic testing. These data suggest that we should be assessing these regions in patients that remain genetically undiagnosed.
An important thing to note, however, is that this does not mean that ¼ of all developmental disease diagnoses are due to variants in non-coding regions. MEF2C is likely a particularly special case. Nethertheless, there are likely other ‘MEF2C like’ genes across different rare diseases where these variants are similarly important.
Diagnoses may be hiding in data that already exists
Another important finding is our ability to detect these variants in the first place. The DDD sequencing data is primarily designed to profile protein-coding regions (the exome). Despite the data being enriched for the exome, however, there are still sequencing reads generated from other (‘off-target’) regions, especially those close to protein-coding regions. We found sufficient sequencing reads to detect genetic variants over 31% of our prioritised UTR bases in these data. This has important implications as the majority of clinical sequencing data that has been generated to date is similarly targeted at protein-coding regions. These data suggest that we might be able to find additional diagnoses in these existing datasets, just through re-analysis.
So where next?
We are still in the early days of understanding the role of non-coding region variants in developmental disorders, and rare disease more broadly. It will be really exciting to uncover additional disease-causing mechanisms as we find more and more examples of functional non-coding region variants. These variants are not only a source of additional genetic diagnoses, but are also an interesting view into gene regulatory mechanisms. In order to do this at scale, we need more datasets that profile the entire genome of both rare disease patients and healthy controls, we need transparent data-sharing across these datasets, and we need both tools and guidelines to support identification, annotation, and clinical interpretation of non-coding variants identified in these individuals.
Some thank yous
This work was a true example of how important collaboration is in science and was a real team effort. Thank you especially to Caroline Wright, Paul Barton, Nick Quaife, Enrique Lara-Pezzi, and Laura Ramos-Hernández - working with you all on this was an absolute pleasure!
Finally, and most importantly, a huge thank you to all of the patients and clinicians involved in the DDD project - without you, none of the ground-breaking research to come out of the study would be possible.